![]()
![]()
Shi-Min YuanI; Hong LinI
DOI: 10.21470/1678-9741-2018-0129
AD = Aortic dissection
Akt = Protein kinase B
CAD = Coronary artery disease
EMT = Epithelial-to-mesenchymal transition
MAPK = Mitogen-activated protein kinase
mRNA = Messenger ribonucleic acid
NmuMG = Normal murine mammary gland
Phosphatidylinositol-3-kinasePI3K =
RNA = Ribonucleic acid
TGF = Transforming growth factor
TGF-ß = Transforming growth factor-beta
TßRI = Transforming growth factor-beta receptor I
TßRII = Transforming growth factor-beta receptor II
INTRODUCTION
Transforming growth factor-beta (TGF-β) is a multi-functional cytokine, which belongs to the TGF superfamily, signaling through transmembrane serine/threonine kinase receptors and Smad proteins. It is usually in a non-activated state and becomes activated when binding to the cell surface receptor, initiating the intracellular signaling transduction. TGF-β plays its biological roles via TGF-β receptors I and II (TβRI and TβRII) on the membranous surface. TGF-β stimulates the production of matrix proteins in vascular smooth muscle cells and matrix protein synthesis, leading to vascular remodeling[1], responsible for aortic aneurysmal formation and possibly coronary artery disorders[2].
Aortic dissection (AD) is the most dangerous cardiovascular disease and it is associated with an extremely high mortality if left without timely diagnosis and treatment[3]. Marfan syndrome is a typical genetic defect with significant propensity for development of AD[4]. It has been noted that endothelial dysfunction of the aortic wall in Marfan patients is caused by fibrillin 1 gene mutations, thus leading to aortic wall fragility and predisposing to AD[5]. In Marfan syndrome, the microfibrillar deficiency would cause inadequate matrix sequestration, with subsequent activation of TGF-β[6]. The increased level of TGF-β is associated with an upregulation of proteases, such as matrix metalloproteinases, and they are responsible for the remodeling process of the aortic wall extracellular matrix[7].
TGF-β is an important cytokine involved in the developmental process of heart and vessels[8]. It is upregulated in relation to injury of vascular walls as a mediator of the fibrotic response, and therefore the upregulation of TGF-β is a result other than a cause of vascular injury[9]. An excessive expression of TGF-β may also hint the progression of secondary disorders[10]. The plasma TGF-β has obtained noteworthy attention in recent years as a reliable biomarker for the evaluation of therapeutic effects of aortic remodeling in Marfan patients[5]. However, the mechanisms that TGF-β signaling regulates during the developmental process of AD and whether TGF-β is increased in aortic conditions in non-Marfan adults remain unknown. Moreover, the evaluation of cytokine expression of TGF-β1 signaling pathways by Western Blotting assays has not been sufficiently described. In order to determine the roles of TGF-β as a potential biomarker of AD development, a prospective study was designed to assess the expression of TGF-β in the aortic tissues of AD patients in comparison to that of patients with coronary artery disease (CAD) receiving coronary artery bypass grafting.
METHODS
Twenty consecutive patients with AD receiving an ascending aorta replacement in this hospital were included in the study group (AD group). Twenty patients with CAD referred to this hospital for coronary artery bypass grafting were taken as controls (CAD group). Totally, 25 patients (including all 20 patients from the AD group and 5 patients from the CAD group) were operated on an urgent basis, and 15 patients of the CAD group received an elective operation. The patients' information is shown in Table 1.
| Group | Aortic dissection | Coronary artery disease | P-value |
|---|---|---|---|
| Case, n | 20 | 20 | NS |
| Gender, male/female, n | 17/3 | 15/5 | NS |
| Age, year | 51.4±8.2 | 58.4±4.6 | NS |
| Smoking, n | 15 | 16 | NS |
| Alcohol, n | 8 | 12 | NS |
| Hypertension, n | 17 | 15 | NS |
| Diabetes mellitus, n | 1 | 10 | NS |
| Renal failure, n | 1 | 2 | NS |
| Operation, n | Aorta replacement, with or without aortic valve replacement (20) |
Off-pump coronary artery bypass (12); on-pump coronary artery bypass (8) |
___ |
| Survival rate, % | 80 | 100 | NS |

Surgically removed aorta specimens from the patients with AD and from the punch holes of the ascending aorta of the patients receiving coronary artery bypass grafting were collected, properly stored, and evaluated quantitatively by Western blotting assay for TGF-β1 and TβRI.
Patients' age and gender did not differ between groups. The aortic tissue specimens were collected immediately after they were severed from the aorta in patients with AD. In patients receiving coronary artery bypass grafting, the tiny aortic tissues measuring 0.2~0.4 cm were taken when the anterior wall of the ascending aorta was punched. The aortic tissues were stored at -80°C, and then thawed for inspection of TGF-β1 and TβRI by Western blotting assay.
Statistics
Quantitative data were presented as mean ± standard deviation with range and median values. The intergroup differences were compared by independent samples t-test. Linear correlations of the relative gray scales between the two groups were assessed. A two-tailed P-value <0.05 was considered statistically significant.
RESULTS
TGF-β1 and TβRI Expressions
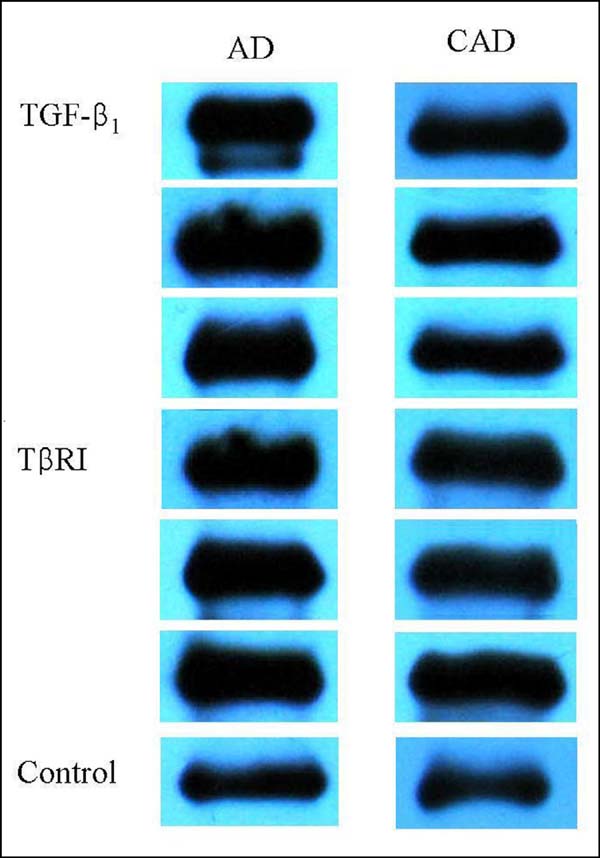
TGF-β1 and TβRI were positively expressed in the aortic tissues of patients of both AD and CAD groups (Figure 1). The TGF-β1 expression in the aortic tissue of AD patients was significantly higher than that of CAD patients. The relative gray scale of the TβRI expression was much higher in AD patients than in CAD patients; however, no significant difference was reached (Figure 2).



Linear Correlations
There was a direct correlation between TGF-β1 and TβRI expressions in the AD group (Y=0.383X + 0.584; r²=0.335, r=0.579, P=0.007); and there was a significant direct correlation between TGF-β1 and TβRI expressions in the CAD group (Y=0.708X + 0.113; r²=0.254, r=0.504, P=0.024) (Figure 3). The coefficient was stronger in AD group than in CAD group.


DISCUSSION
The present study revealed that TGF-β1 and TβRI were positively expressed in the aortic tissues of AD and CAD patients, with a direct correlationship between TGF-β1 and TβRI expressions in patients of AD and CAD groups. The results illustrated that the aortic wall remodeling happened in response to atherosclerotic or dissecting insults, and the close relationships between them revealed the mutual biological effects between the ligands and the receptors. The disparity of the biomarkers' expressions between the two groups warranted another approach for the understanding of possible differences in aortic remodeling fashions between groups in terms of nature, mechanism, mode, and activities of the TGF signaling.
The expressions of cytokines of the TGF-β signaling pathway depend on the E3 ubiquitin-protein ligases Smurf1 and Smurf2. Knockdown of endogenous Smurf1 or Smurf2 by ribonucleic acid (RNA) interference significantly suppressed the anti-inflammatory effects of TGF-β1 [11]. TGF-β is a potent regulator of vascular development and remodeling, contributing to atherosclerotic process and restenosis by augmenting neointimal proliferation and collagen accumulation[12]. TGF-β could activate the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt) pathway in normal murine mammary gland (NmuMG) epithelial cells 30 minutes after treatment, and the activation reached a peak value at 2 hours[13]. It could enhance the activities of cyclin dependent kinase inhibitors, p15Ink4B and p21Waf1/Cip1[14,15], thereby influencing the sensitivity of cell proliferation and apoptosis. TGF-β activates p38 mitogen-activated protein kinase (MAPK), which is essential for TGF-β-induced apoptosis and for epithelial-to-mesenchymal transition (EMT)[16].
TGF-β induces extracellular matrix synthesis and prevents enzymatic breakdown of the extracellular matrix by activating Smad and upregulating the expressions of fibronectin and connective tissue growth factor, etc.[17]. Other pro-fibrotic factors, such as angiotensin II and advanced glycation end products, might also activate the Smad signaling pathway via TGF-β-dependent or independent mechanisms[18]. The Smads show diverse functions based on different family members. In vascular smooth muscle cells, the overexpression of Smad7 inhibits the expressions of fibronectin and connective tissue growth factor, whereas Smad2/3 and Smad4 are for deposition of the extracellular matrix mediated by TGF-β. Alternatively, Smad2/3 signaling pathway could be activated in a TGF-β dependent manner 24 hours after stimulating vascular smooth muscle cells by angiotensin II[19].
TGF-β has been identified as a key anti-atherogenic agent due to its fibrosis-stimulating effects. TβRI is usually expressed in unstable plaques, whereas TβRII is expressed in normal vessels. A study showed that TβRI increased by two folds 8 hours after vascular injury, TGF-β1 increased by ten folds within 24 hours, and TβRII increased by three folds[20]. Experimental atherosclerosis was observed to be accelerated when TGF-β1 was suppressed[21]. However, in advanced atherosclerosis, TGF-β1 may behave as a proatherogenic stimulus by increasing extracellular matrix formation and fibrosis[22]. In such occasions, TGF-β1 was likely to lose its anti-atherogenic effects[23,24]. Thus, in humans with atherosclerosis, decreased TGF-β1 signaling and loss of p27 expression could be found[25]. Attenuated TGF-β1 signaling and expressions might be an attribution of vascular aging caused by atherosclerosis[26].
Enhanced TGF-β signaling transduction and weakened receptor kinase were found in patients with thoracic aortic aneurysm[27]. Jones et al.[27] discovered TGF-β signaling transduction dysfunction in the mouse model of thoracic aorta aneurysm, and they revealed the inherent close relationships between the expression of TGF-β/Smads and the phenotype of vascular smooth muscle cells. TGF-β/Smads play a role in molecular regulation in the reversion of vascular smooth muscle cells, from a synthetic phenotype to a differentiated phenotype[28]. An experiment on injured carotid arterial smooth muscle cells of Sprague-Dawley rats with the use of reverse transcription-polymerase chain reaction and Western blotting assays showed that TGF-β1 stimulated smooth muscle cell proliferations in a concentration-dependent manner. The expressions of messenger ribonucleic acid (mRNA) and proteins of TGF-β1 and TβRII were much higher in the injured carotid artery than in the control group. However, no difference was found in the expressions of TβRI mRNA and protein between the two groups. It illustrated that after vascular injury, the phenotype of the smooth muscle cells changed with unusual proliferations, leading to increased synthesis and secretion of TGF-β1. The results verified the functions of TGF-β1 in promoting proliferation of the injured smooth muscle cells depending on various receptor subtypes[29].
It has been reported that TGF-β1 was unevenly distributed in the aortic wall of patients with AD and of heart donors: it was expressed the highest in the media, followed by a higher expression in the intima. Intergroup comparisons revealed that TGF-β1 expression in the entire aortic wall, media, and adventitia were much higher in patients with AD than in heart donor control subjects[30]. Transgenic mouse models of Marfan syndrome also disclosed a key role of increased TGF-β signaling in promoting vascular remodeling, dilation, and aneurysmal formation[31]. Therefore, TGF-β plays a protective role in controlling excessive activations of monocytes and macrophages, inhibiting matrix degradation, and promoting survival of the smooth muscle cells of the aortic media[32].
When TGF-β/Smad signaling pathway is interfered with TβRI, angiogenesis and production of matrix metalloproteinases may be reduced, whereas the non-Smad dependent pathway depends on TβRI, and may enhance the fibrinolytic function of the extracellular matrix[27]. It was demonstrated that TGF-β may have anti-inflammatory and fibrosis-promoting effects, and may also prevent unstable plaque rupture[33]. The inhibitory effect of TβRII to TGF-β may promote fibrosis and reduce tissue inflammation[34]. By using neutralizing antibodies of TGF-β1, β2, and β3 to block the signaling transduction, there would be an association with an accelerated atherosclerosis and an unstable plaque phenotype[35]. Although TGF-β1 can inhibit proliferation, it may enhance early immigration of the injured tissue in the mesenchymal cells by increasing the production of matrix metalloproteinase-1[36]. Animal experiments on Marfan models showed that activation of TGF-β and the concurrent upregulation of matrix metalloproteinases seemed to contribute to aortic aneurysm formation[37]. Pathologically, AD is typically characterized by medial degeneration, smooth muscle cell depletion, and extracellular matrix degradation[38].
CONCLUSION
The differential expressions of cytokines of TGF-β signaling pathways supported the hypotheses of diverse vascular remodeling fashions between AD and CAD. The aortic remodeling of AD might differ from that of CAD concerning the nature, mechanism, mode, and activities of TGF signaling pathway. The vascular remodeling might be a degradation of the extracellular matrix by the upregulation of matrix metalloproteinases in the aorta of AD patients, while it might be a deposition of the extracellular matrix in the aorta of CAD patients. The development of AD could be associated with a significantly enhanced function of TGF-β/Smad signaling transduction as a result of aortic remodeling incorporating both vascular injury and repair.
REFERENCES
1. Dai JH. New progress of transforming growth factor-ß. J MedRes. 2007;36(1):103-4.
2. Ye P, Chen W, Wu J, Huang X, Li J, Wang S, et al. GM-CSF contributesto aortic aneurysms resulting from SMAD3 deficiency. J Clin Invest.2013;123(5):2317-31.
3. Weigang E, Nienaber CA, Rehders TC, Ince H, Vahl CF, Beyersdorf F.Management of patients with aortic dissection. Dtsch Arztebl Int.2008;105(38):639-45.
4. Westaby S. Aortic dissection in Marfan's syndrome. Ann Thorac Surg.1999;67(6):1861-3.
5. Benke K, Ágg B, Szilveszter B, Tarr F, Nagy ZB, Pólos M, et al. Therole of transforming growth factor-beta in Marfan syndrome. Cardiol J.2013;20(3):227-34.
6. Dietz HC. 2006 Curt Stern Award Address. Marfan syndrome: frommolecules to medicines. Am J Hum Genet. 2007;81(4):662-7.
7. Booms P, Pregla R, Ney A, Barthel F, Reinhardt DP, Pletschacher A,et al. RGD-containing fibrillin-1 fragments upregulate matrix metalloproteinaseexpression in cell culture: a potential factor in the pathogenesis of the Marfansyndrome. Hum Genet. 2005;116(1-2):51-61. [MedLine]
8. Sridurongrit S, Larsson J, Schwartz R, Ruiz-Lozano P, Kaartinen V.Signaling via the TGF-beta type I receptor Alk5 in heart development. Dev Biol.2008;322(1):208-18.
9. Franken R, Radonic T, den Hartog AW, Groenink M, Pals G, van Eijk M,et al; COMPARE study group. The revised role of TGF-ß in aortic aneurysmsin Marfan syndrome. Neth Heart J. 2015;23(2):116-21.
10. Matt P, Schoenhoff F, Habashi J, Holm T, Van Erp C, Loch D, et al;GenTAC Consortium. Circulating transforming growth factor-beta in Marfansyndrome. Circulation. 2009;120(6):526-32.
11. Lee YS, Park JS, Kim JH, Jung SM, Lee JY, Kim SJ, et al.Smad6-specific recruitment of Smurf E3 ligases mediates TGF-ß1-induceddegradation of MyD88 in TLR4 signalling. Nat Commun.2011;2:460.
12. Bobik A. Transforming growth factor-betas and vascular disorders.Arterioscler Thromb Vasc Biol. 2006;26(8):1712-20.
13. Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL.Phosphatidylinositol 3-kinase function is required for transforming growthfactor ß-mediated epithelial to mesenchymal 1transition and cellmigration. J Biol Chem. 2000;275(47):36803-10.
14. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM.Direct binding of Smad3 and Smad4 to critical TGFß-inducible elements inthe promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J.1998;17(11):3091-100.
15. Wakefield LM, Roberts AB. TGF-ß signaling: positive andnegative effects on tumorigenesis. Curr Opin Genet Dev.2002;12(1):22-9.
16. Yu L, Hébert MC, Zhang YE. TGF-ß receptor-activated p38 MAPkinase mediates Smad-independent TGF-ß responses. EMBO J.2002;21(14):3749-59.
17. Ihn H. Pathogenesis of fibrosis: role of TGF-ß and CTGF. CurrOpin Rheumatol. 2002;14(6):681-5.
18. Lan HY. Diverse roles of TGF-ß/Smads in renal fibrosis andinflammation. Int J Biol Sci. 2011;7(7):1056-67.
19. Feng XH, Lin X, Derynck R. Smad2, Smad3 and Smad4 cooperate with Sp1to induce p15(Ink4B) transcription in response to TGF-ß. EMBO J.2000;19(19):5178-93.
20. Chen WB, Lenschow W, Tiede K, Fischer JW, Kalthoff H, Ungefroren H.Smad4/DPC4-dependent regulation of biglycan gene expression by transforminggrowth factor-ß in pancreatic tumor cells. J Biol Chem.2002;277(39):36118-28.
21. Lutgens E, Gijbels M, Smook M, Heeringa P, Gotwals P, KotelianskyVE, et al. Transforming growth factor-ß mediates balance betweeninflammation and fibrosis during plaque progression. Arterioscler Thromb VascBiol. 2002;22(6):975-82.
22. Strippoli R, Benedicto I, Perez Lozano ML, Pellinen T, Sandoval P,Lopez-Cabrera M, et al. Inhibition of transforming growth factor-activatedkinase 1 (TAK1) blocks and reverses epithelial to mesenchymal transition ofmesothelial cells. PLoS One. 2012;7(2):e31492. [MedLine]
23. Redondo S, Navarro-Dorado J, Ramajo M, Medina Ú, Tejerina T. Thecomplex regulation of TGF-ß in cardiovascular disease. Vasc Health RiskManag. 2012;8:533-9.
24. Grainger DJ. Transforming growth factor ß andatherosclerosis: so far, so good for the protective cytokine hypothesis.Arterioscler Thromb Vasc Biol. 2004;24(3):399-404. [MedLine]
25. Ihling C, Technau K, Gross V, Schulte-Mönting J, Zeiher AM, SchaeferHE. Concordant upregulation of type II-TGF-ß-receptor, thecyclin-dependent kinases inhibitor P27Kip1 and cyclin E in human atherosclerotictissue: implications for lesion cellularity. Atherosclerosis.1999;144(1):7-14. [MedLine]
26. Redondo S, Navarro-Dorado J, Ramajo M, Medina Ú, Molina-Sanchez P,Garces Z, et al. Age-dependent defective TGF-beta1 signaling in patientsundergoing coronary artery bypass grafting. J Cardiothorac Surg.2014;9:24.
27. Jones JA, Spinale FG, Ikonomidis JS. Transforming growth factor-betasignaling in thoracic aortic aneurysm development: a paradox in pathogenesis. JVasc Res. 2009;46(2):119-37.
28. Yan CH, Shen JL, Yan J, Wang BQ, Zhe W, Zhang GY, et al.Proliferation of vascular smooth muscle cell and the expression of transforminggrowth factor ß/Smads. Chin J Clin Rehabil.2005;9(19):50-2.
29. Lin ZH, Wu KG, Li GS, Xie LD, Lin PH. Role of transforming growthfactor ß1 and its receptor in the intimal hyperplasia after vascularinjury. Chin J Gerontol. 2015;35(8):2200-2.
30. Yang SG, Wang CS, Chen H, Zhu SJ, Hong T, Lai H. TGF-ß1expression and distribution in the extracellular matrix of the dissected wall ofthoracic aorta. Chin J Thorac Cardiovasc Surg. 2010:26(1):33-6.
31. Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, et al.Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfansyndrome. Science. 2006;312(5770):117-21.
32. Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S,et al. TGF-ß activity protects against inflammatory aortic aneurysmprogression and complications in angiotensin II-infused mice. J Clin Invest.2010;120(2):422-32.
33. Hobson KG, Cho K, Adamson LK, Greenhalgh DG. Burn-induced thymicapoptosis corresponds with altered TGF-ß1 and Smad 2/3. J Surg Res.2002;105(1):4-9. [MedLine]
34. Xu J, Oakley J, McGee EA. Stage-specific expression of Smad2 andSmad3 during folliculogenesis. Biol Reprod. 2002;66(6):1571-8.
35. Robertson AK, Rudling M, Zhou X, Gorelik L, Flavell RA, Hansson GK.Disruption of TGF-ß signaling in T cells accelerates atherosclerosis. JClin Invest. 2003;112(9):1342-50.
36. Gugjoo MB, Amarpal, Abdelbaset-Ismail A, Aithal HP, Kinjavdekar P,Pawde AM, et al. Mesenchymal stem cells with IGF-1 and TGF- ß1 in laminingel for osteochondral defects in rabbits. Biomed Pharmacother.2017;93:1165-74. [MedLine]
37. Xiong W, Meisinger T, Knispel R, Worth JM, Baxter BT. MMP-2regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. CircRes. 2012;110(12):e92-e101. [MedLine]
38. Wu D, Shen YH, Russell L, Coselli JS, LeMaire SA. Molecularmechanisms of thoracic aortic dissection. J Surg Res2013;184(2):907-24.
No financial support.
No conflict of interest.
Authors' roles & responsibilities
SMY Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; final approval of the version to be published
HL Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; final approval of the version to be published
Article receive on Wednesday, May 2, 2018
 All scientific articles published at www.bjcvs.org are licensed under a Creative Commons license
All scientific articles published at www.bjcvs.org are licensed under a Creative Commons license













All rights reserved 2017 / © 2024 Brazilian Society of Cardiovascular Surgery
DEVELOPMENT BY ![]()
 Read in English
Read in English
 English PDF
English PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket